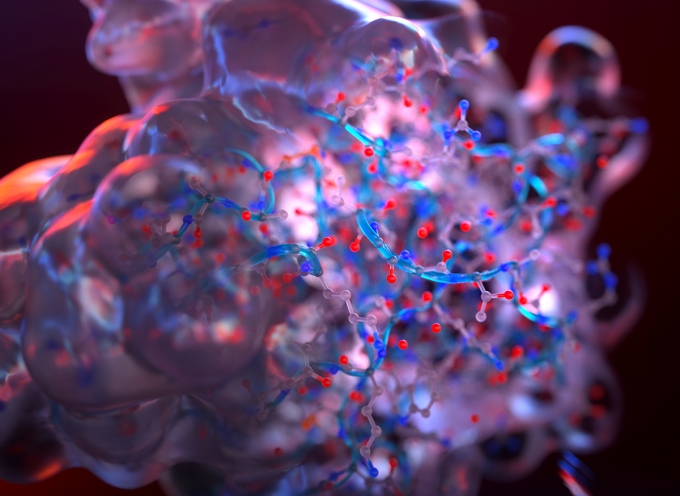
Scientists rely heavily on simulations to understand the nature and behavior of molecular systems. Professor M. Scott Shell, in the Department of Chemical Engineering, develops computer simulations to describe how atoms and molecules generate forces that cause them to attract or repel each other and, thus, determine how they rotate, move, and evolve over time. That collective activity gives rise to the properties of everyday materials like water and allows biological molecules to perform their unique and complex functions. Simulations provide a virtual microscope to understand how such complexity emerges, and in turn, how the same biologically inspired behaviors can be used to create novel synthetic materials for a wide range of applications.
Computer simulations provide a detailed picture of every atom in every molecule, as long as the system is small enough. “Sometimes you need only a thousand- or ten-thousand-atom simulation, as in liquid water, because if you simulate more than that, the system starts to look like a sea of the same without providing any new information,” Shell explains.
“Even with supercomputers, computing the activity of a million atoms is a heroic kind of simulation. And there are millions or billions of atoms in many systems we want to study.”
But real limits to computing power, and the extensive time required to calculate the dynamic forces between atoms, constrain the ability to simulate systems having millions or billions of atoms.
“For every atom, the program must remember its location and speed, and calculate its interactions with all other atoms [and it must do this repeatedly for incredibly small increments of time], so you quickly start running out of computing power,” Shell explains. “Even with supercomputers, computing the activity of a million atoms is a heroic kind of simulation.”
As an example, Shell says that in a month of 24/7 computing time using the most advanced hardware, it is normally possible to simulate the atomic activity that occurs during only about a millionth of a second in a billionth of a billionth of a liter of water.
That level of simulation, as incomplete as it may seem, turns out to be sufficient for understanding a wide range of molecular properties, but many more systems of interest — particularly complex molecules that perform biological functions — could be characterized if those limits could be expanded.
Shell went to work on that challenge and for his efforts received the American Institute of Chemical Engineers Computational Molecular Science and Engineering Forum Impact Award in 2017. He was recognized for developing algorithms that allow researchers to move well past the traditional limits of computational modeling and simulate molecular structures and systems that are significantly more complex. The work is based on a theoretical concept called “relative entropy,” for which his group is now well known.
“I’m extremely happy about this award, especially because it comes from a community of people who know my work well and therefore can actually evaluate it in great detail,” Shell said of the recognition. “When I began as an assistant professor, this idea for relative entropy theory was new and, perhaps, risky. There were three to four years when I didn’t know if it was something people would care about.”
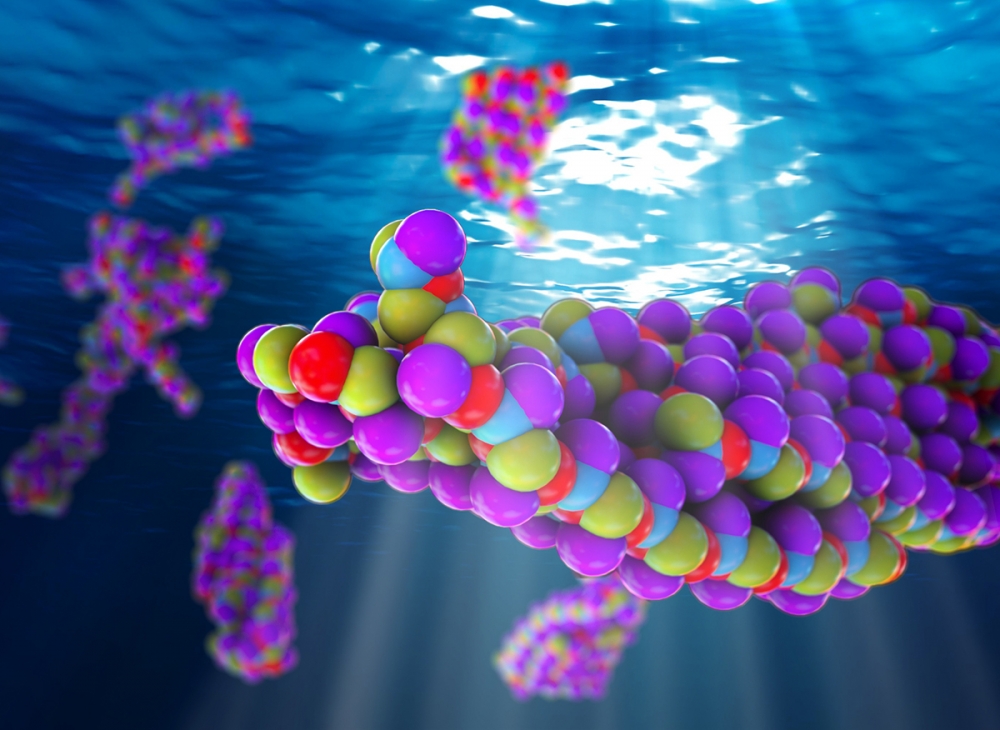
An Artist's interpretation of peptides composed of molecular groups in specific patterns self-assembling into functional structures
Early Research
When Shell arrived at UCSB in 2007, the field of peptides — biological polymer chains of two or more amino acids — was burgeoning. “Unlike conventional synthetic polymers, such as plastics, you can arrange the sequence of molecular groups along a peptide in a precise pattern,” Shell says. “That’s how nature does it.”
He noted that the ability to control sequences enables substantial control over behavior, because when peptides composed of a particular pattern of molecular groups are dissolved in water, as in the human body, they self-assemble into structures whose shape, properties, and functionality are determined by that pattern. In recent years, efforts have emerged to create biological molecules that are like peptides and have novel sequences that leverage those unique self-assembly properties to program-in new structures.
In the early 2000s, researchers discovered special patterns of synthetic peptides that, when dissolved in water, self-assemble to form nanomaterials of various shapes — hollow tubes, square platelets, fibers, and spheres, for example. “The systems self-assemble based on the precise patterning that balances all the interactions,” Shell explains. “Understanding the relationship between the sequence patterns and the resulting structures would offer insight into how to engineer nanomaterials precisely.”
As an example, he adds, “Say I’ve made a peptide sequence and wonder what nanomaterial shape it will form in solution, or, even more desirable but significantly more challenging, I want a specific shape and wonder what sequence I should use to achieve it.
Tackling that experimentally through trial and error is a really long process, so a computational method for understanding what sequences become which structures would be valuable and establish what engineers refer to as ‘design rules.’
“There wasn’t a good theory for how all that worked,” he continues, “and since chemical engineers think about how to predict and understand soft materials that are driven by thermodynamics and self-assembly processes, and since I had just completed a postdoc in protein-folding simulations, I thought that working to understand some of the underlying design principles would allow us to develop theory to advance the field.”
Given limitations of computing power, Shell realized that to simulate large-scale processes like those associated with self-assembled peptide systems or even the natural molecular processes inside cells would require a new approach. He developed a strategy based on the idea that it’s not necessary to keep track of every atom in a system. His theory allows for a process called “coarse-graining,” in which only the most important atomic motions are identified and simulated, eliminating the need to compute the behavior of millions of individual atoms, without sacrificing accuracy. The secret was figuring out how to get rid of unnecessary details, and a key discovery was realizing that he would need to measure exactly how much information is lost when removing atoms to simplify a system for simulation.

Shell explains: “When a system is coarse-grained, inevitably some information is lost, because you no longer know where all the atoms are. It turns out that the loss can be quantified as a number. The higher the number, the more severe the loss and the more potentially inaccurate the coarse-graining approximation becomes.”
He realized that relative entropy was the correct way to measure information loss in coarse-grained molecular simulations. Eventually, he was able to create algorithms that minimized relative entropy and, thus, information loss. He could then design accurate coarse-grained models that made it far easier to simulate complex systems.
“That was our hook,” Shell recalls. “We had this thermodynamic perspective that allowed us to simplify systems automatically so that we didn’t have to model the behavior of every atom, and we could therefore achieve significantly more thorough simulations of complicated systems without sacrificing fidelity or realism.” Those approaches, plus other simulation techniques developed by Shell and his students, have broadened understanding of synthetic peptide materials.